


In this article, we provide a quick introduction to adsorption science isotherms. We begin with a derivation of the Gibbs adsorption equation. Then, we introduce two of the most common adsorption isotherm models: the Langmuir isotherm and the BET isotherm. Each section is accompanied by an example problem.
1. The Gibbs adsorption equation (Shaw, 1992; Pashley & Karaman, 2004; Cosgrove, 2005)
The Gibbs adsorption equation enables the extent of adsorption at a liquid surface to be estimated from surface tension data. Before deriving this expression, it is important to realize the limitations associated with this model – and, for that matter, all of the adsorption isotherms studied in this article. It is convenient to regard the interface between two phases as a mathematical plane, such as SS in Figure 1. This approach, however, is unrealistic, especially if an adsorbed film is present. Not only will such a film have a certain thickness, but also its presence may influence the nearby structure (for example, by dipole-dipole interaction, especially in an aqueous phase) and result in an interfacial region of varying composition with an appreciable thickness in terms of molecular dimensions. Nevertheless, theories based on discrete layer separation offer a good degree of accuracy relatively to experimental measurements. One of the earliest such models, proposed by the legendary American thermodynamicist, Josiah Gibbs, is briefly described below.
Figure 1. Interfaces in ideal and real systems.
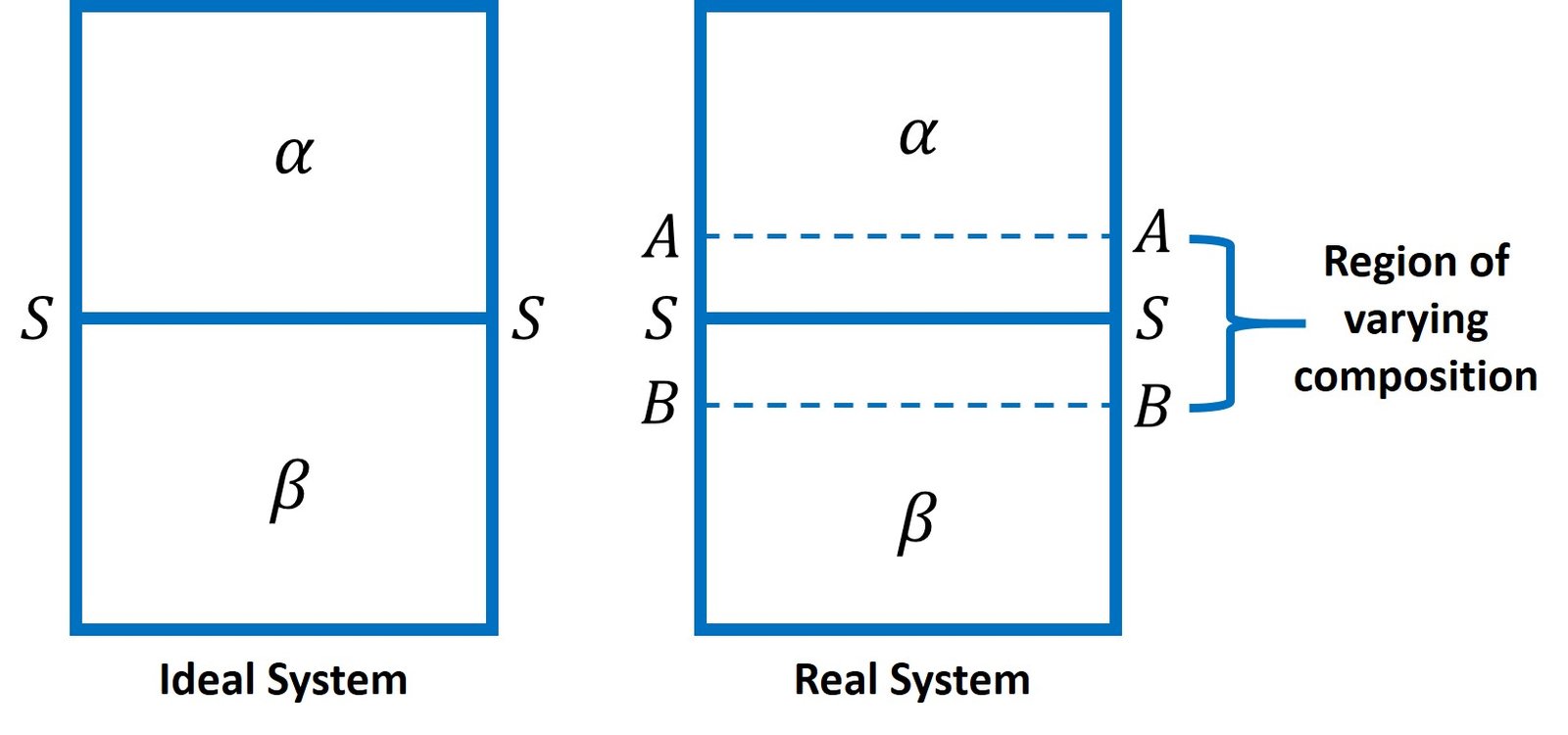
If a mathematical plane is indeed taken to represent the interface between the two phases, adsorption can be described conveniently in terms of surface excess concentrations. Let us consider the interface between two phases, say between a liquid and a vapor, where a solute (i) is dissolved in the liquid phase. When the solute increases in concentration near the interface between the two phases, there must be a surface excess of solute 

where A is interfacial area. Bear in mind that 
Let us now examine the effect of adsorption on the interfacial energy (



or, in the case of several components,

The change in
Figure 2. Change in surface energy caused by addition of a solute.

Gibbs’ equation can be readily extended to a solution of two components, in which case we may write
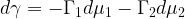
and hence the adsorption excess for one of the components is given by

Thus, in principle, we could determine the adsorption excess of one of the components from surface tension measurements, if we could vary 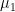



Now, since
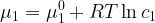
differentiation with respect to concentration 

so that, combining this result with the expression for 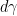
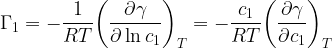
This relation is the Gibbs adsorption isotherm in its usual formulation. Experimental measurements of 

It is important to bear in mind that the Gibbs equation in the form above applies to non-dissociating materials (e.g., non-ionic surfactants). For ionic surfactants of the form 

If no electrolyte is added, electroneutrality of the surface requires that 


Cosgrove (2005) recommends using the equation above with the mean ionic activity 

Example 1
For a 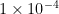

Note that 

The surface area available for each molecule is then
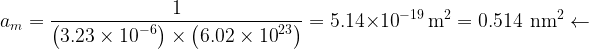
2. Adsorption isotherms – the Langmuir isotherm (Modified from Tien, 2019)
The Langmuir isotherm is perhaps the most widely used isotherm expression for representing physical adsorption equilibrium data. Although it may be derived in several different ways, the kinetic approach is used in most textbooks. The basic assumptions are:
1. Adsorption of adsorbates takes place at well-defined sites.
2. All adsorption sites are the same (energetically), and each site accommodates only one adsorbate molecule.
3. There is no lateral interaction between adsorbed molecules.
The adsorption rate is assumed to be

where 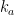
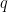



where 


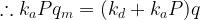


Let 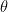

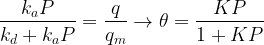
This is the basic form of the Langmuir adsorption isotherm.
Example 2
At 273 K and 10 bar, the Langmuir adsorption of a gas on a solid surface gave the fraction of surface coverage as 0.01. In this case, what is the value of the Langmuir adsorption constant?
From the Langmuir isotherm equation, we have
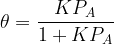
We were given the fraction of surface coverage 




3. Adsorption isotherms – the Brunauer-Emmet-Teller (BET) isotherm (Ghosh, 2009)
The theory of chemical adsorption proposed by Langmuir is based on the formation of a monolayer. However, multilayers can form in physical adsorption. The isotherm in the case of a monolayer assumes the shape shown as Type I in Figure 3. The extent of adsorption increases with pressure and ultimately reaches a limiting value, as predicted by the Langmuir theory. However, for adsorption involving multilayer formation at low temperatures, at least four other types of adsorption isotherm can be observed; see below. (In the graphs shown, y denotes the amount of gas adsorbed by m kg of adsorbent at pressure P.)
Figure 3. Adsorption isotherms.

To explain such varied adsorption isotherms, Brunauer, Emmet and Teller proposed, in 1938, a theory of multilayer adsorption. They posited that, even after the formation of the monolayer, other layers of gas may condense on it. Langmuir’s idea of fixed adsorption sites was, however, retained. They suggested that there is equilibrium between the first layer and the adsorbent, and similar dynamic criteria were assumed to exist for the successive molecular layers. The Van der Waals forces provide the binding energy in these successive layers. The equation derived by them is

where 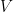


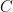
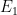


From the BET equation, it is clear that a plot of 
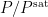
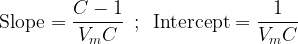
Accordingly,

Thus, the BET equation permits us to extract from multilayer adsorption data the volume of adsorbed gas that would saturate the surface if the adsorption were limited to a monolayer. The most immediate application of this quantity is the calculation of specific surface area. The volume of gas adsorbed when the surface is covered by a monolayer, 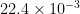
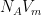



Here,
Example 3
The following data were obtained for adsorption of nitrogen on 0.88 kg of nitrogen-activated alumina. The data are adapted from Tien (2019).
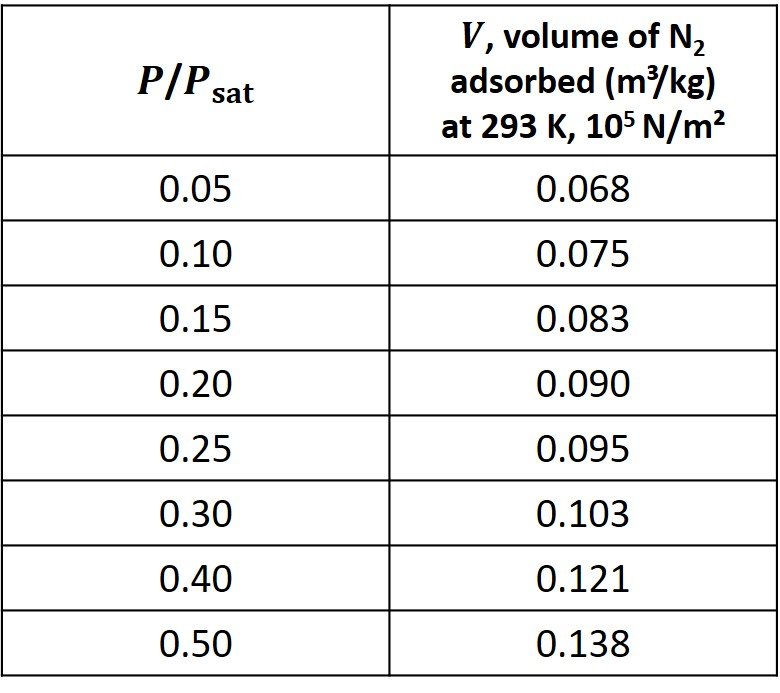
A) Estimate the volume of nitrogen adsorbed over 1 kg of alumina when a monolayer is formed.
B) Calculate the specific surface area of this alumina sample. The area occupied by one nitrogen molecule is 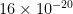
According to the BET model, a plot of 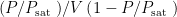

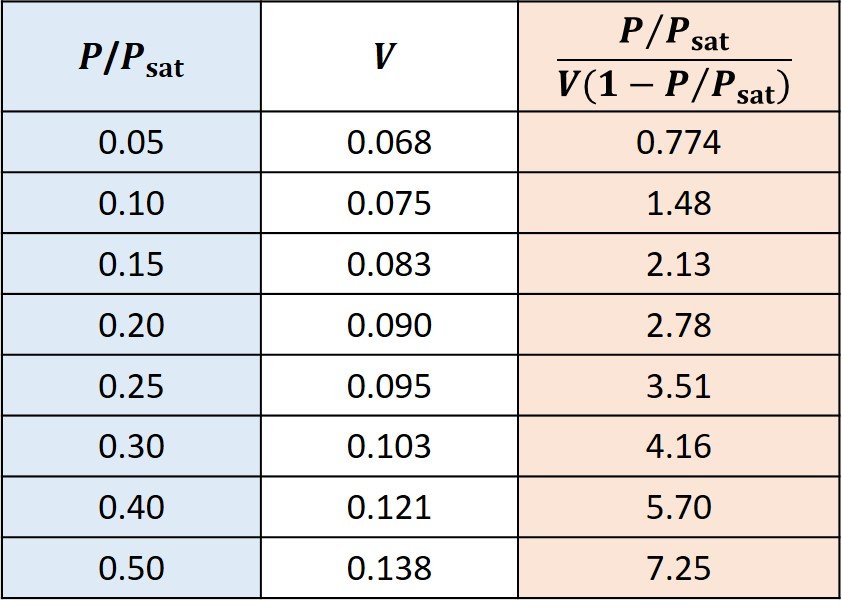
Then, the data in the red column are plotted against the data in the blue column, as shown.
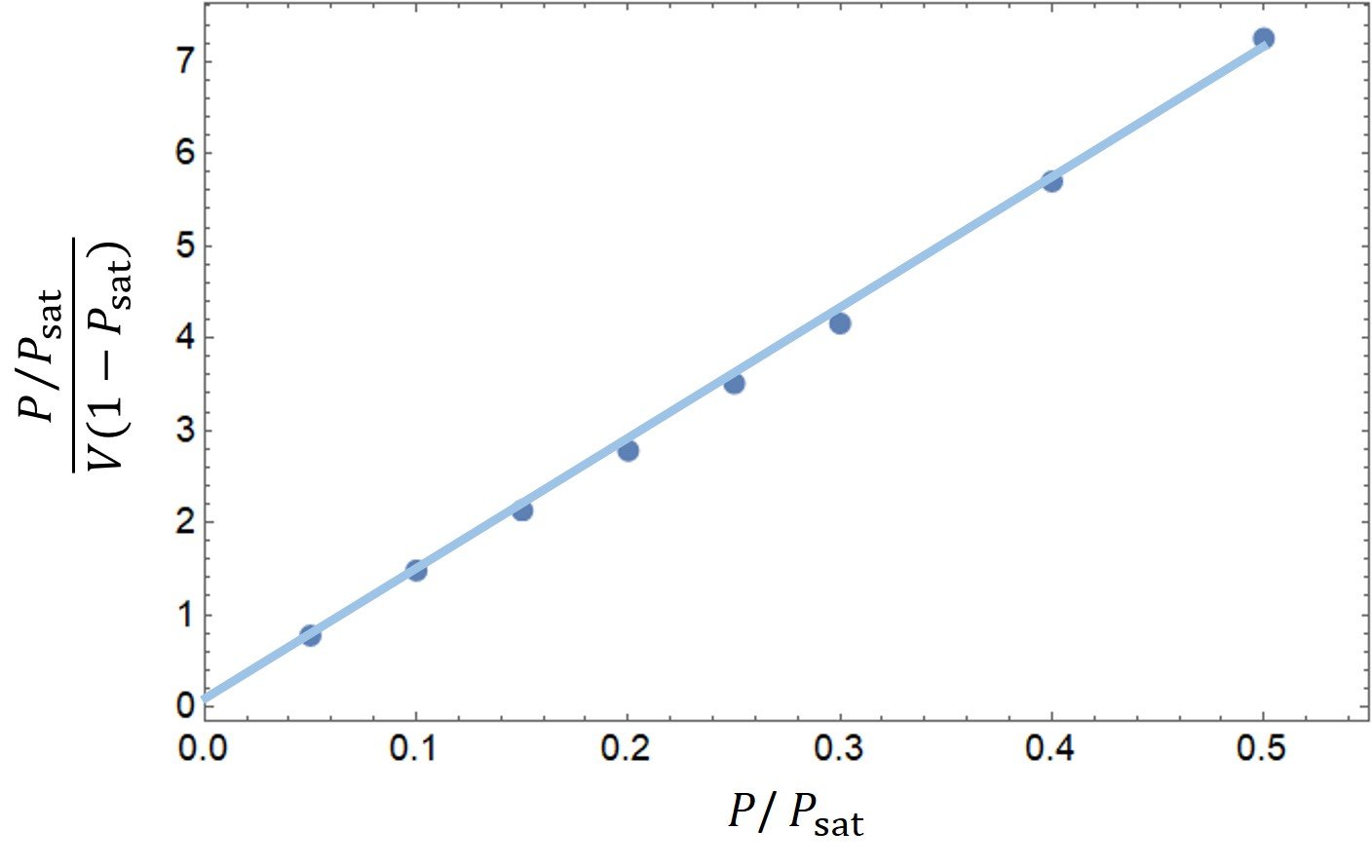
Clearly, there is a linear trend in this dataset. Fitting these data to a first-degree equation yields a line with slope

and intercept
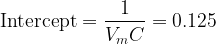
Solving both equations simultaneously gives
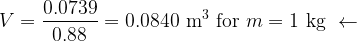
Lastly, the specific surface area is

References
• COSGROVE, T. (Ed.) (2005). Colloid Science: Principles, Methods and Applications. Oxford: Blackwell Publishing.
• GHOSH, P. (2009). Colloid and Surface Science. New Delhi: PHI Learning.
• PASHLEY, R. and KARAMAN, M. (2004). Applied Colloid and Surface Chemistry. Hoboken: John Wiley and Sons.
• SHAW, D. (1992). Introduction to Colloid and Surface Chemistry. 4th edition. Oxford: Butterworth-Heinemann.
• TIEN, C. (2019). Introduction to Adsorption. Amsterdam: Elsevier.

